Desde el año 2008, el día 28 de febrero se conmemora el Día Mundial de las Enfermedades Raras con el propósito de crear conciencia y ayudar a todas las personas que padecen alguna enfermedad de las denominadas "raras", para que reciban el debido diagnóstico y tratamiento y de esta forma que puedan llevar una vida mejor. Se escogió esta fecha porque el mes de febrero es muy particular, "raro", pues unos años tiene 28 días y otros 29. Por lo tanto, el Día Mundial de las Enfermedades Raras, se celebra cada 28 o 29 de febrero, dependiendo si estamos, o no, en un año bisiesto.
El Dr. Lluis Montoliu apuntaba que la traducción del inglés "rare" a "raras" ha sido muy poco afortunada. "Rare diseases" se debería haber traducido como "enfermedades poco frecuentes". El adjetivo "rara" podría ser hiriente, negativo, e incluso discriminar y separar a las personas afectadas, y a sus familias. Realmente a las enfermedades raras las deberíamos llamar enfermedades de baja prevalencia, pues son patologías que afectan a menos de 5 personas cada 10.000 habitantes.
La Federación Española de Enfermedades Raras (FEDER) indica en su página web que se estima que existen más de 7000 enfermedades raras, de las cuales 6.172 han sido ya identificadas, según datos de Orphanet. Cada enfermedad rara es muy variable en cuanto a prevalencia. Existen enfermedades raras más comunes, como es el caso de la fibrosis quística, el trastorno genético más común entre los niños caucásicos, que afecta por lo menos a 1 de cada 8.000 personas. En otros casos la población afectada por una enfermedad rara es mucho menor. Por ejemplo en la acromatopsia o ceguera de colores (los individuos no perciben colores, solo tonalidades de gris) la frecuencia estimada en la población es de 1-9 de cada 100.000 personas. El 80% de las enfermedades raras tienen un origen genético, en algunos casos aún desconocido. La dificultad para ofrecer un diagnóstico certero y la escasez de tratamientos son problemas comunes en estas patologías.
En el IIBM hay once grupos que trabajan en enfermedades raras. Colaboran con grupos de investigación de hospitales para desarrollar ensayos funcionales y genéticos, así como para buscar nuevos tratamientos, es decir la investigación que se lleva a cabo en el IIBM tiene un fuerte componente traslacional. Muy importante también es la cooperación con las asociaciones de pacientes que están muy implicadas en el trabajo que desarrollan los investigadores.
A continuación, resumimos el trabajo que llevan a cabo los grupos del IIBM en enfermedades raras.
Grupo de Enfermedades Asociadas a Autofagia. Dres. Ricardo Escalante Hernández y Oliver Vincent
El grupo de los Dres. Escalante y Vincent estudian las bases moleculares de enfermedades raras relacionadas con el proceso de la autofagia. Actualmente trabajan en BPAN (del inglés: Beta Propeller-associated neurodegeneration) y ChAC (del inglés: Chorea-acanthocytosis), dos enfermedades neurodegenerativas sin tratamiento efectivo. Utilizan microorganismos modelo y líneas celulares humanas para entender la función molecular y celular de los genes implicados en estas enfermedades y caracterizar el efecto de mutaciones identificadas en pacientes. Si queréis conocer más podéis leer su página web
Grupo de Mecanismos Moleculares de la Fisiopatología Mitocondrial. Dr. Miguel Fernández Moreno
Las enfermedades mitocondriales (MD) son aquellas provocadas por alteraciones funcionales del sistema OXPHOS, formado por los cuatro complejos de la cadena respiratoria (Complejos I-IV) más la ATP sintasa o Complejo V. Aunque individualmente son consideradas enfermedades raras, las MD en conjunto constituyen el grupo más numeroso de errores congénitos del metabolismo. Las MD son genética y clínicamente muy heterogéneas, y se manifiestan con un único síntoma razonablemente moderado como sordera o intolerancia al ejercicio, o en forma de síndromes devastadores incompatibles con la vida. Estas enfermedades pueden ser consecuencia de mutaciones en genes nucleares o mitocondriales. El grupo del Dr. Fernández Moreno tiene varios objetivos: la identificación de genes no descritos implicados en la función de OXPHOS, la caracterización del mecanismo de acción molecular de un grupo reducido de proteínas implicadas de un modo singular en la síntesis de tRNAs mitocondriales, el estudio de la relación del ADN mitocondrial y la tumorogénesis y el desarrollo de modelos animales para el estudio de las enfermedades mitocondriales.
Grupo de Ciliopatías. Dr. Francesc García Gonzalo
Las ciliopatías son enfermedades genéticas raras causadas por disfunción de cilios, finas protrusiones de la membrana celular formadas por microtúbulos y que pueden funcionar como motores o sensores. La mayoría de ciliopatías afectan a los cilios primarios, antenas celulares que detectan señales ópticas, mecánicas o químicas, según el tipo celular. Para funcionar correctamente, dichas antenas deben primero sintonizarse, es decir, los cilios deben acumular los receptores y mediadores que les permitirán captar las señales apropiadas. En el laboratorio del Dr. García Gonzalo, estudian los mecanismos moleculares por los que las células sintonizan sus cilios, así como los procesos patogénicos que interfieren con dicha sintonización en ciliopatías como los síndromes de Joubert o Bardet-Biedl, cuyos síntomas incluyen, entre otros, déficits cognitivos y motores, ceguera, quistes renales, obesidad y polidactilia.
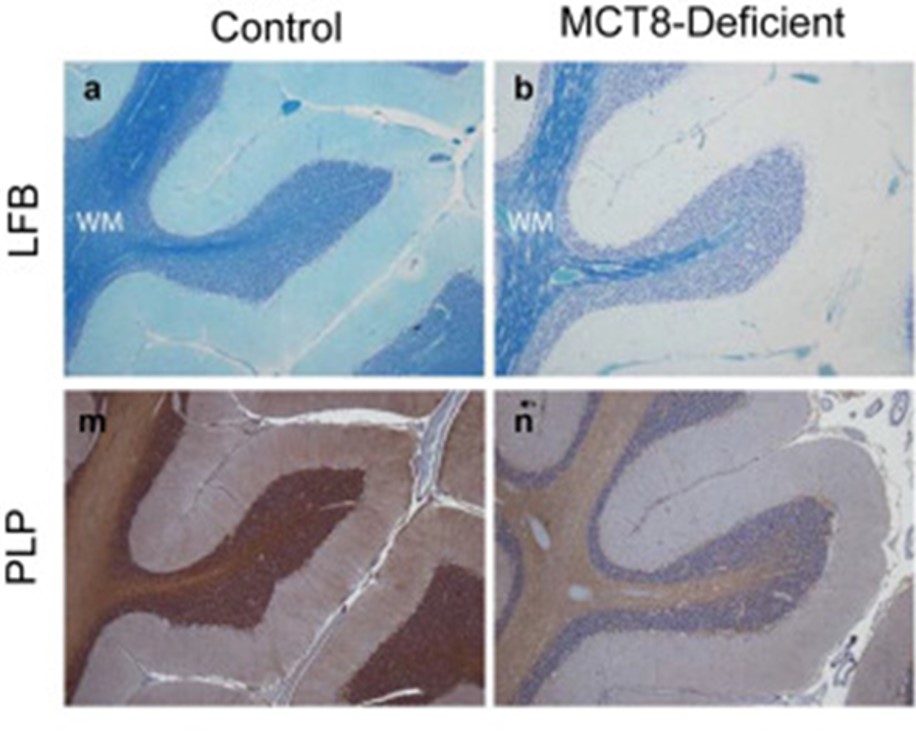
Grupo de Hormonas Tiroideas y Sistema Nervioso Central. Dra. Ana Guadaño Ferraz
El grupo de la Dra. Ana Guadaño (@ThyroidLabIIBM) trabaja en el estudio del síndrome de Allan-Herndon-Dudley o deficiencia de MCT8, una enfermedad rara debida a mutaciones en el transportador de hormonas tiroideas MCT8. Su grupo tiene como objetivos la identificación de biomarcadores para la fisiopatología cerebral de los pacientes afectados por esta patología, así como el desarrollo de estrategias terapéuticas en ratones modelo de la enfermedad para prevenir o mejorar las alteraciones neurológicas.
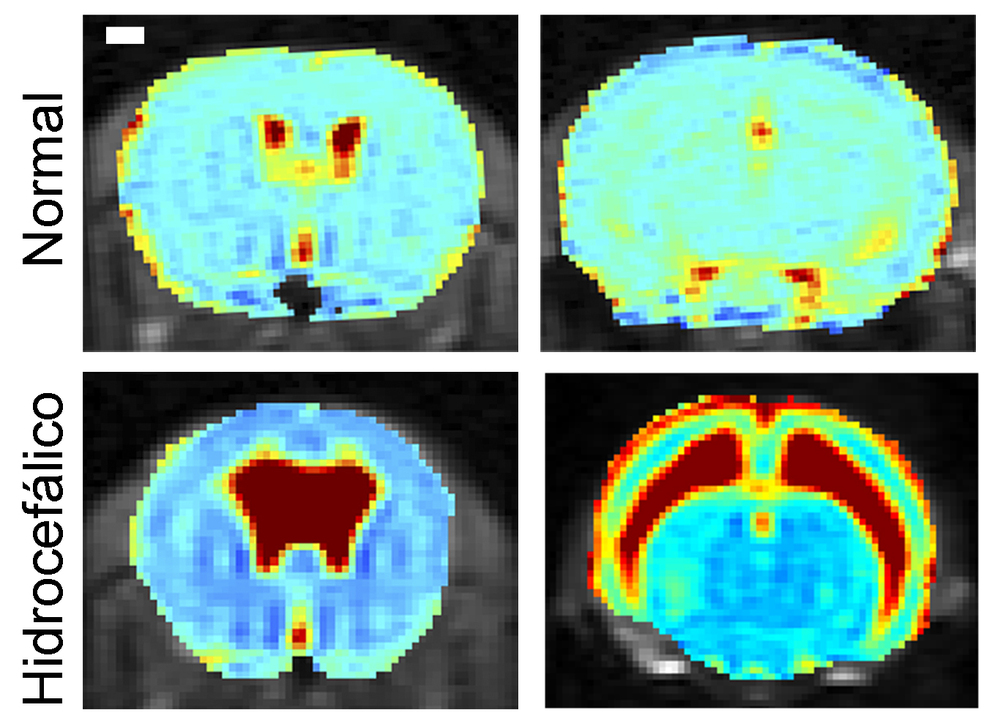
Grupo de Nuevas Dianas en Neurodegeneración y Neuroprotección. Dra. Teresa Iglesias Vacas
La Dra. Teresa Iglesias clonó el gen KIDINS220, y con su equipo ha estado investigando la función de este gen en distintas neuropatologías. Recientemente se han identificado variantes patogénicas de KIDINS220 que producen un síndrome infantil denominado SINO (del inglés: Spastic paraplegia, Intellectual disability, Nystagmus and Obesity). Se trata de una enfermedad neurológica rara asociada a un retraso del desarrollo, paraplejia espástica que hace necesario el uso de silla de ruedas, discapacidad intelectual, alteraciones oftalmológicas y obesidad. El síndrome SINO cursa además con una acumulación de líquido en el cerebro o hidrocefalia. Su grupo ha descubierto que los ratones deficientes en Kidins220 también presentan hidrocefalia, identificando además los mecanismos implicados en esta acumulación de líquido. Actualmente, utilizan modelos experimentales con la finalidad de desarrollar terapias para los pacientes afectados con esta enfermedad.
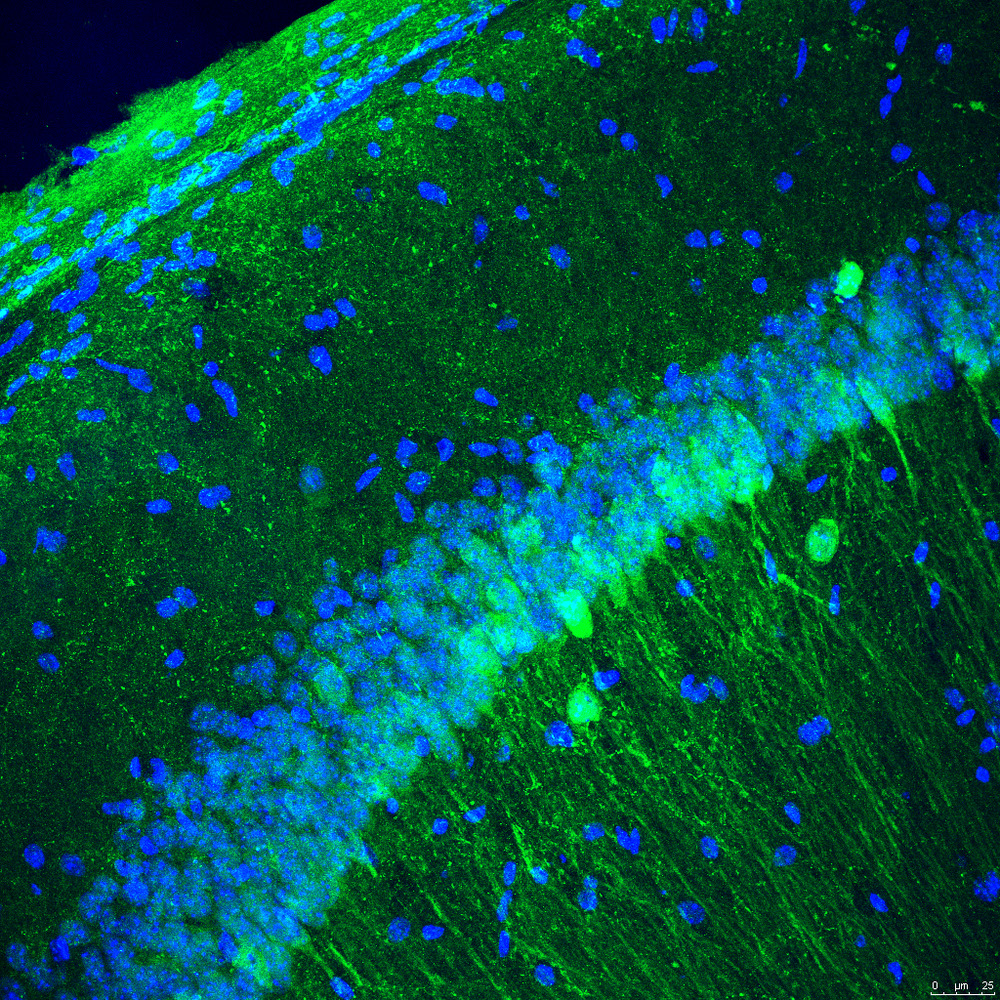
Grupo de Parkinson, ELA y Tauopatías: Nuevas Perspectivas (Cure4Brain). Dra. Isabel Lastres-Becker
La esclerosis lateral amiotrófica (ELA) y la demencia frontotemporal (DFT), también denominada degeneración lobar frontotemporal (DLFT), son dos enfermedades neurodegenerativas raras intrínsecamente asociadas que se superponen a nivel clínico, neuropatológico y, sobre todo, genético. Uno de los grandes retos del laboratorio de la Dra. Lastres-Becker es descubrir nuevas dianas terapéuticas, cuya modulación farmacológica pueda frenar o detener los procesos degenerativos en estas enfermedades tan devastadoras.
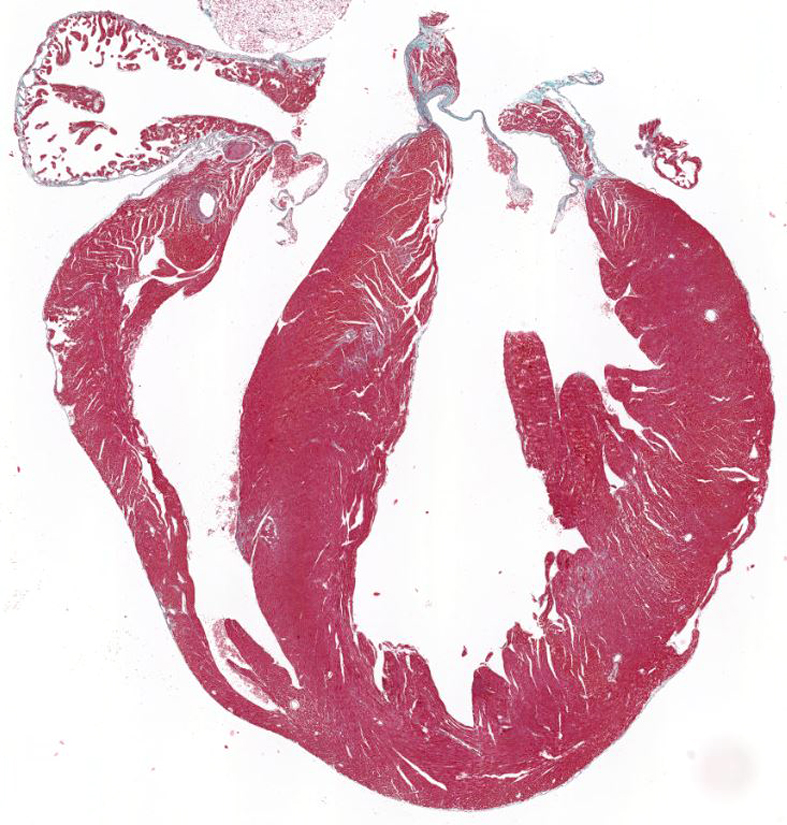
Grupo de Homeostasis del Oxígeno en el Sistema Cardiovascular. Dra. Silvia Martín Puig
El grupo de la Dra. Martín Puig investiga los mecanismos moleculares y los condicionantes genéticos implicados en la aparición de la enfermedad de Kawasaki (EK), una vasculitis pediátrica de origen desconocido y principal causa de enfermedad cardiovascular adquirida en la infancia. Un porcentaje de pacientes no tratados a tiempo, o con resistencia a los actuales tratamientos clínicos, desarrolla alteraciones cardiovasculares severas, entre las que destacan las dilataciones y aneurismas coronarias, trombosis, inflamación cardiaca y eventualmente infarto de miocardio. Este grupo ha desarrollado un modelo animal que recapitula estas alteraciones e investigan como la vía de hipoxia interviene en la patogénesis vascular e inflamatoria de la EK. Además, están intentando identificar nuevas variantes genéticas que puedan explicar el mayor riesgo de desarrollo de aneurismas coronarias o resistencia al tratamiento mediante técnicas de secuenciación masiva. El grupo también investiga las bases moleculares y condicionantes genéticos de una nueva enfermedad rara, el síndrome Multisistémico Inflamatorio en Niños (MISC) que es una manifestación clínica severa asociada a la infección pediátrica por SARS-CoV2 y con sintomatología solapante a la EK.
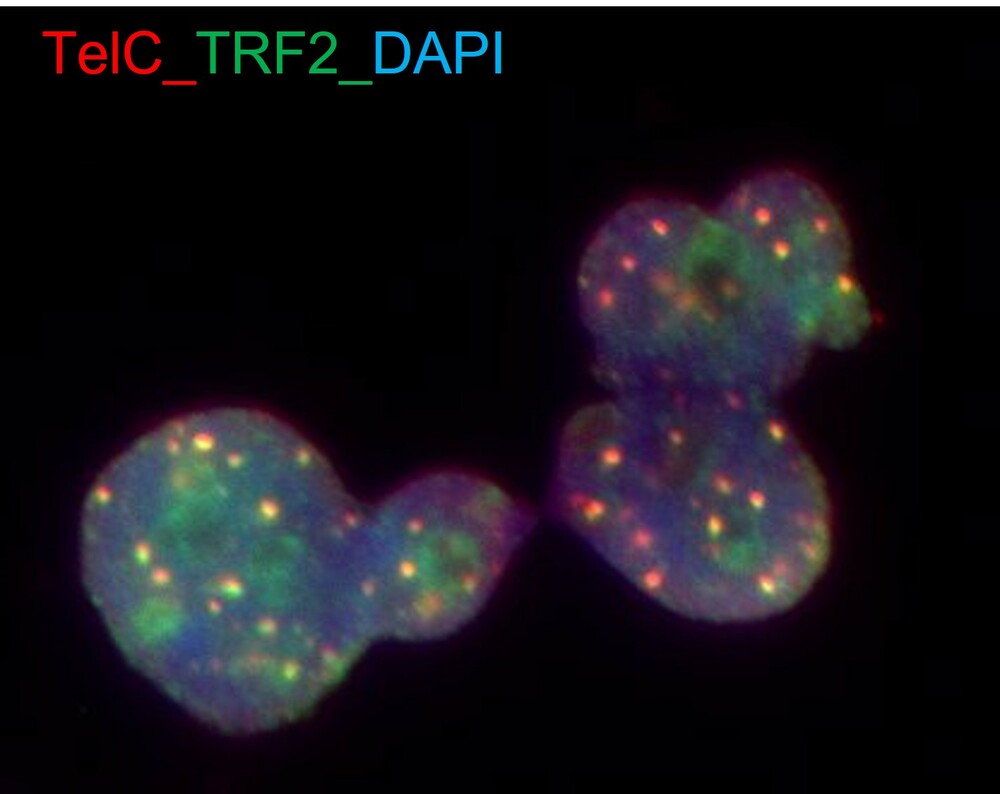
Grupo de Enfermedades Teloméricas y Terapias Experimentales. Dres. Rosario Perona Abellón, Rosa Guerrero-López y Leandro Sastre Garzón
El grupo coordinado por el Dr. Leandro Sastre trabaja en enfermedades relacionadas con el acortamiento de los telómeros, incluyendo disqueratosis congénita y fibrosis pulmonar idiopática. Estas patologías son enfermedades de envejecimiento acelerado de la piel, la medula ósea y otros órganos. Las manifestaciones más graves son anemia, inmunoinsuficiencia y fibrosis pulmonar, las cuales suponen una amenaza importante para la vida de los pacientes. Este grupo trabaja en el diagnóstico de la enfermedad y en la búsqueda de nuevos tratamientos que corrijan la ausencia de tratamientos curativos que existe en la actualidad. La Dra. Rosa Guerrero-López también investiga la identificación de factores genéticos implicados en la patogenia y la susceptibilidad para el desarrollo de la encefalitis de Rasmussen, una enfermedad autoinmune-inflamatoria del sistema nervioso central, en el marco de un proyecto colaborativo liderado por el Hospital Infantil Niño Jesús.
Grupo de Genética y Mecanismos Fisiopatológicos de Anomalías Congénitas. Dr. Víctor Luis Ruiz-Pérez
El grupo del Dr.Ruiz-Pérez tiene una larga experiencia en la investigación de enfermedades raras que cursan con alteraciones del desarrollo con especial énfasis en las ciliopatías esqueléticas y síndromes asociados a fragilidad ósea. En sus investigaciones utilizan una metodología que combina técnicas de genética, como el análisis de variantes procedentes de la secuenciación de exomas, con el estudio de modelos celulares o animales adecuados a cada patología. Los principales objetivos de su investigación son la identificación de nuevos genes causantes de trastornos del desarrollo, contribuyendo así a mejorar el diagnóstico de estas enfermedades, y la determinación de los mecanismos celulares y moleculares implicados en cada caso, un conocimiento que es imprescindible para el desarrollo de nuevas terapias.
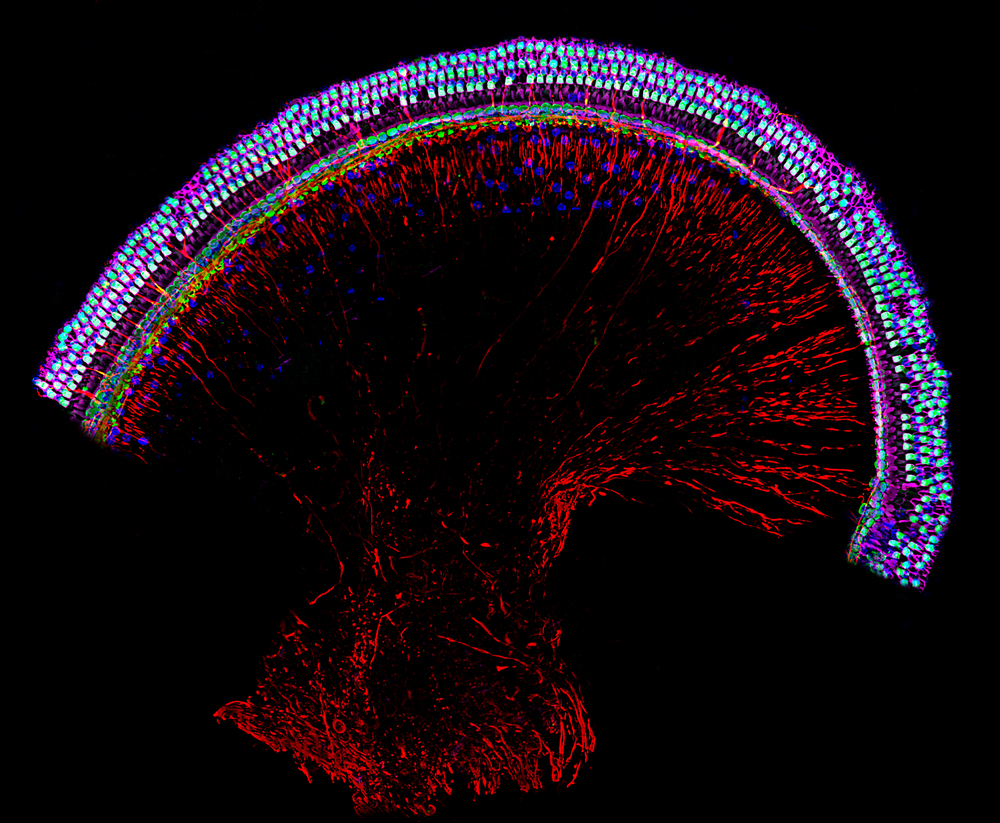
Grupo de Neuropatología Auditiva y Mielinopatías. Dres. Isabel Varela Nieto, Silvia Murillo Cuesta, Ana María Jiménez Lara y José Miguel Cosgaya Manrique
El laboratorio coordinado por la Dra. Isabel Varela trabaja en el estudio de la pérdida auditiva y las mielinopatías. Entre las causas de la pérdida auditiva existen las de etiología genética y las adquiridas, incluyendo el envejecimiento auditivo precoz y la hipoacusia por exposición a ruido, entre otras, las cuales también tienen un fuerte componente de predisposición genética. Muchas de estas hipoacusias son enfermedades raras por su baja prevalencia individual. El grupo también estudia los schwannomas, unas mielinopatías tumorales raras que pueden afectar al nervio auditivovestibular y producir hipoacusia. Sus objetivos son comprender las bases genéticas, moleculares y celulares de estas enfermedades, desarrollar nuevas estrategias terapéuticas y llevar a cabo ensayos preclínicos en diversos modelos experimentales para evaluar su eficacia.
Grupo de Modelos Preclínicos y Nuevas Terapias. Dr. Juan Manuel Zapata Hernández
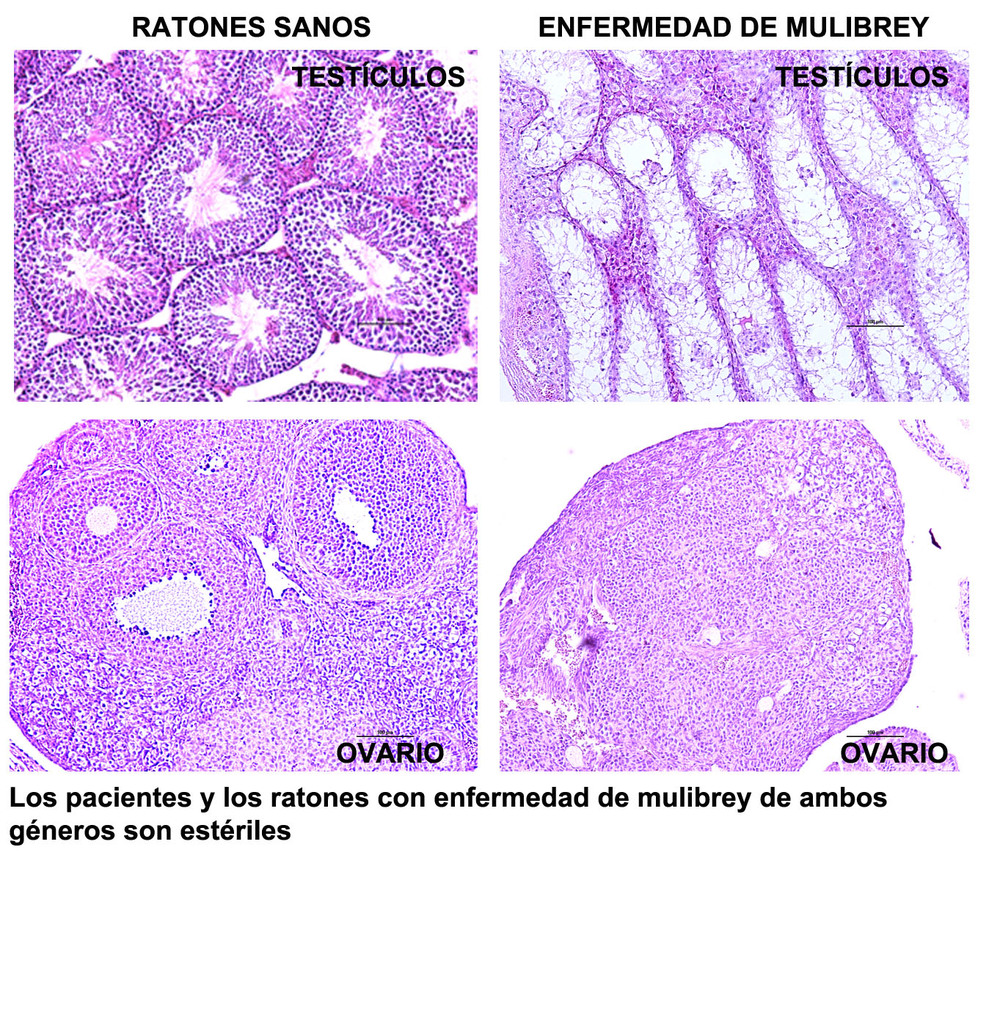
El grupo del Dr. Zapata investigan dos enfermedades raras, la leucemia linfática crónica (LLC) y el trastorno del crecimiento MULIBREY o síndrome de Perheentupa. La LLC es una enfermedad aún incurable. Generan modelos murinos de esta leucemia para entender su etiología y evolución, y buscan nuevos fármacos efectivos contra células de pacientes resistentes a los tratamientos actuales. El trastorno del crecimiento MULIBREY, causado por mutaciones en el gen TRIM37, es una enfermedad extremadamente rara caracterizada por restricción del crecimiento y otras manifestaciones multiorgánicas. Este grupo estudia el papel de TRIM37 en la regulación de la respuesta inmunitaria innata en células no linfoides a través de modelos celulares y murinos.